ARTICLES

Gene causing rare heart and lung disease identified Genomewide screening, animal studies used in long-term research on pulmonary arterial hypertension
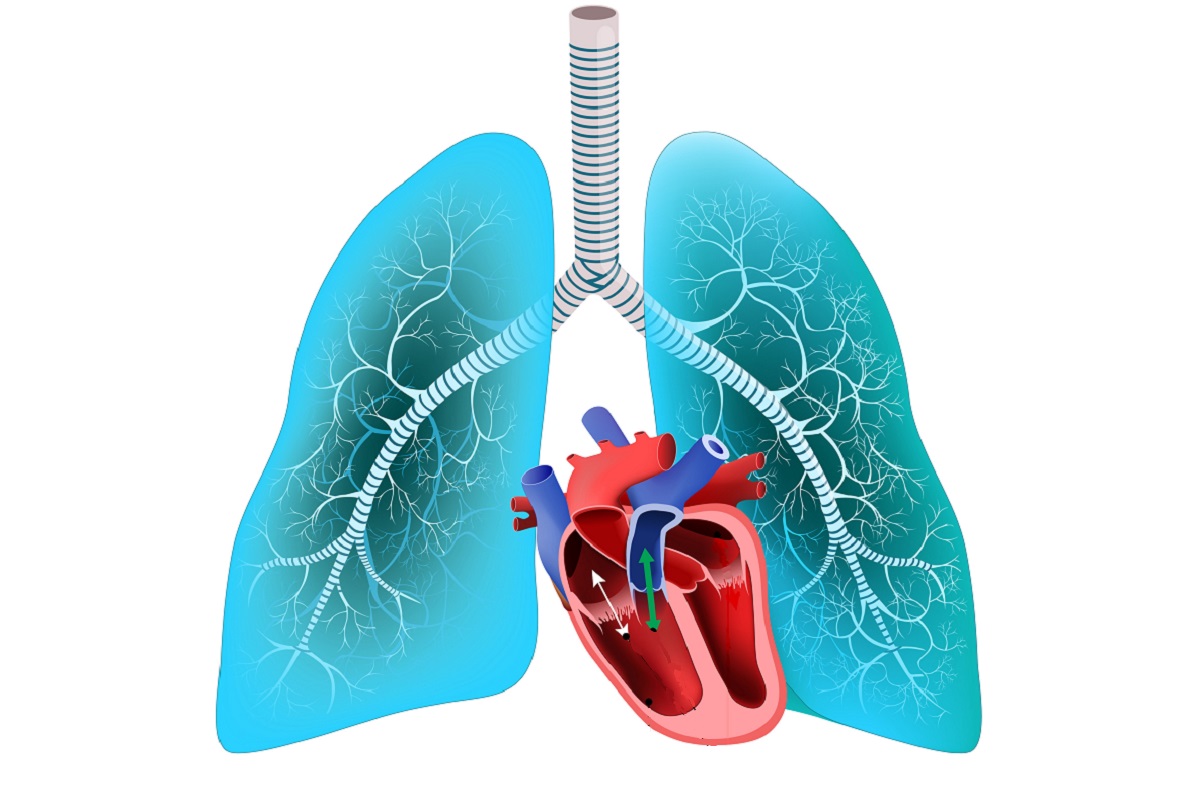
Pulmonary arterial hypertension (PAH) is a form of pulmonary hypertension, a type of high blood pressure in the lungs. In PAH, small arteries in the lungs become narrowed and damaged, which increases resistance to blood flow through the lungs. To overcome the greater resistance, blood pressure increases and the heart needs to work harder to pump blood through the lungs, eventually causing damage to the heart. In a long-term international study, researchers identified a gene in mice that may counteract the cause of this rare disease. © Designua/Shutterstock.com
Researchers have identified a gene in mice that may counteract the cause of a rare and progressive type of heart disease, pulmonary arterial hypertension. The result comes after more than a decade of work by experts in Japan and Sweden.
Pulmonary hypertension is a type of high blood pressure in the lungs and the right side of the heart. In pulmonary arterial hypertension (PAH), small arteries in the lungs become narrowed and damaged, which increases the resistance to blood flow through the lungs. To overcome the greater resistance, blood pressure increases in the arteries of the lung and right heart (right side of the heart, or right ventricle). The heart needs to work harder to pump blood through the lungs for gas exchange, which eventually causes damage to the heart. Patients with PAH experience symptoms like excessive fatigue or fainting, and the extra stress on the heart can cause death.
Although many cases of PAH have no known cause, some cases are genetically inherited. PAH is listed as a rare and intractable disease by the American National Organization for Rare Disorders, and there are only 1-2 cases of the disorder per 1 million people in Japan, according to the Japan Intractable Diseases Information Center.
Previous research had revealed that patients with inherited cases of PAH tend to have mutations in the BMPR2 gene, which encodes a receptor protein on the cell surface that binds to bone morphogenetic protein (BMP). In addition, mutations have been identified in several other genes related to the BMP signaling pathway. BMP uses transcription factor SMAD proteins to convert and relay signals through pathways in the cells, and regulates the expression of target genes in the nucleus.
"Since mutations in those factors can cause pulmonary arterial hypertension, we wanted to know what is the target gene regulated by BMP/SMAD," said Dr. Kohei Miyazono, M.D., Ph.D., from the University of Tokyo Graduate School of Medicine and last author of the research paper published in Science Signaling.
Researchers began the study by searching for genes that were directly regulated by the SMAD proteins in the nucleus. Using a result from next-generation sequencing, they saw that SMAD could directly regulate the expression of the ATOH8 protein in endothelial cells — flat cells constituting the inner lining of blood and lymph vessels.
To study the biological relevance, they used zebrafish and mice that lacked normal ATOH8 activity as animal models of human disease. They found that mice lacking the Atoh8 gene had symptoms resembling human PAH, which supports the idea that ATOH8 is necessary to prevent PAH.
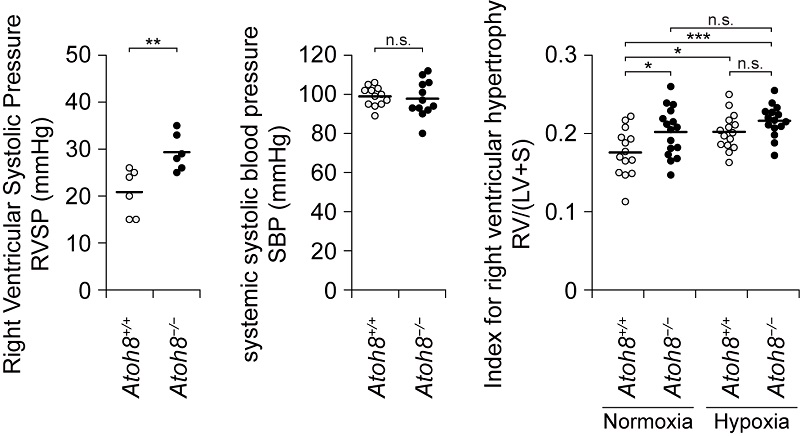
Atoh8-deficient mice exhibit a phenotype resembling human PAH. (Left) Assessment of right ventricular systolic pressure (RVSP) and systemic systolic blood pressure (SBP) of Atoh8-deficient mice. (Right) Assessment of right ventricular hypertrophy of Atoh8-deficient mice. © 2020 Morikawa, et al.
"Fortunately, we have good collaborators in Japan and in Sweden. We were all happy to receive the result that Atoh8-deficient mice had higher right ventricular systolic pressure, implicating ATOH8 in the pathogenesis of PAH," said Miyazono.
Results of additional laboratory experiments using cultured endothelial cells showed that ATOH8 could counteract signals of low oxygen levels (hypoxia). One of those signaling molecules is hypoxia-inducible factor (HIF), whose activity can be stopped by BMP and ATOH8. These findings suggest that the mechanism observed in the cultured cells may explain how BMP and ATOH8 prevent the development of PAH in mice and humans.
"Since inhibitors for HIF have been developed and are under clinical trials as anticancer drugs, our results suggest that those HIF inhibitors may be useful in the treatment of PAH," said Miyazono.
This research project is the result of an international collaboration by UTokyo with scientists at Uppsala University in Sweden, and Mie University and Kyoto University in Japan. It was supported by KAKENHI (Grants-in-Aid for Scientific Research) for Innovative Area on Integrated Analysis and Regulation of Cellular Diversity and the Global Center of Excellence Program (Integrative Life Science Based on the Study of Biosignaling Mechanisms).
Papers
Masato Morikawa, Yoshihide Mitani, Katarina Holmborn, Taichi Kato, Daizo Koinuma, Junko Maruyama, Eleftheria Vasilaki, Hirofumi Sawada, Mai Kobayashi, Takayuki Ozawa, Yasuyuki Morishita, Yasumasa Bessho, Shingo Maeda, Johan Ledin, Hiroyuki Aburatani, Ryoichiro Kageyama, Kazuo Maruyama, Carl-Henrik Heldin, and Kohei Miyazono, "The ALK-1/SMAD/ATOH8 axis attenuates hypoxic responses and protects against the development of pulmonary arterial hypertension," Science Signaling: November 12, 2019, doi:10.1126/scisignal.aay4430.
Link (Publication , UTokyo Repository)
, UTokyo Repository)
Related links
- Miyazono Laboratory
- Department of Pathology, Immunology and Microbiology
- Graduate School of Medicine
- Read about this research in Japanese